A Moth in the Tree of Life at Sanger
The life of a sample at the Tree of Life labs at the Wellcome Sanger Institute starts with an email forewarning us, for example, of the imminent arrival of carefully identified moth specimens from Wytham Woods in barcoded freezer vials. On the day, an email from stores summons Nancy from her desk to collect the freezer parcel, and she scans the vials, checks them against the detailed sample manifest and places them in the -80°C freezer. Most samples are then passed onto the Sanger Samples Management Facility, a carefully backed-up rank of freezers that holds not just the Tree of Life samples but thousands upon thousands of samples from other Sanger programmes in human genetics, cancer, cellular genetics, pathogens and microbes.

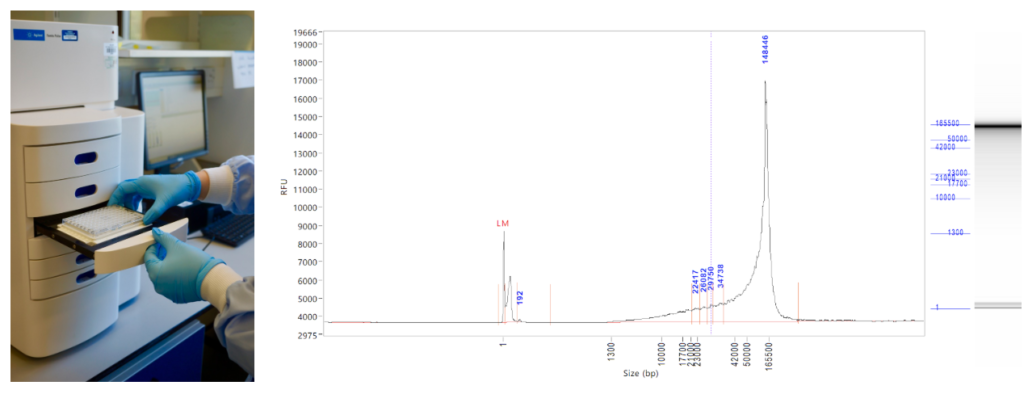
There the moth sample waits in the freezers for a short time while Nancy compiles the instructions for sequencing: Is the moth especially rare? What DNA extraction method should be used? How big is the genome likely to be and thus how much data do we need to generate? The sample is then processed to retrieve very long DNA, either by the Tree of Life lab team, or our colleagues in Sanger’s Scientific Operations. For example, Radka (from the Tree of Life lab team of Radka, Michelle, Clare, Robin and Harriet) might take the moth sample and pulverise it before digesting the protein and extracting the DNA. She will check the quality of the DNA samples using a FemtoPulse instrument, which uses very little sample (a blessing when the sample is very small) to accurately quantify and size fragments up to 165 kilobases (kb). We have extraction methods that work well for moths and beetles and mammals and flies, and we are improving the quality of extractions from plants and fungi.
Good quality DNA then moves into library production. Making a large-insert library for the Pacific Biosciences SEQUEL II instrument or the Oxford Nanopore Promethion instrument is part art and part routine. As with extractions we currently share the load of library production between the Tree of Life team and Scientific Operations. For the moth, Radka will take some of the DNA, shear it to just the right length (usually between 13-18kb) and perform the molecular biology steps that are needed to prepare it for sequencing.
The library is handed over to the Scientific Operations Long Read team to load onto the big machines, the SEQUEL II and Promethion sequencers. These technologies have changed what is possible in genomics, and are the basis of the confidence that we can generate genomes from our thousands of target species. The machines take from 24 hrs to 3 days to run, producing tens of gigabases of raw data from each library. For the moth, we will need only one run of one of the sequencers to generate enough data for primary assembly.
Meanwhile, Mike and Matt in Scientific Operations prepare some special long-range sequencing libraries from unsheared DNA and remaining sample. 10X Genomics linked read cloud libraries generate data that allow us to jump over and resolve complicated repeats in the moth genome. Hi-C libraries capture the three dimensional arrangement of chromosomes in each nucleus of the moth, sampling DNA fragments that are close to each other in 3D space, but far apart on the linear, stretched-out chromosome. These 10X and Hi-C libraries generate data sets that are used to link long-read data into chromosomes. 10X and Hi-C data are generated on the fleet of Illumina sequencing instruments in Scientific Operations.

The SciOps team checks the data are of good quality, parks them on the Sanger’s (very) large hard drive system, and sends an email announcing the availability of another species’-worth of data.
Shane’s email inbox fills with messages about completed sequencing runs, and when all the moth’s data are ready he and his Tree of Life Assembly team (Marcela and Ksenia) kick off the process of assembly on the Sanger’s compute farm. This uses cutting edge software to identify overlapping long reads, disentangle confusions that result from repeats and errors, and finally stitch everything together first of all into contigs (stretches of contiguous AGCT sequence) and then into scaffolds (contigs that are ordered and oriented using long-range data). Only five years ago we would have struggled to generate assemblies with mean contig lengths over 50 kb. With the long read Pacific Biosciences and Oxford Nanopore data we now get assemblies with mean contig lengths over 1 Megabase (Mb), frequently over 5 Mb and sometimes over 10 Mb. For species like our moth, which has a genome of 600 Mb, once Shane adds the 10X and Hi-C data, these assemblies fall into chromosomes.
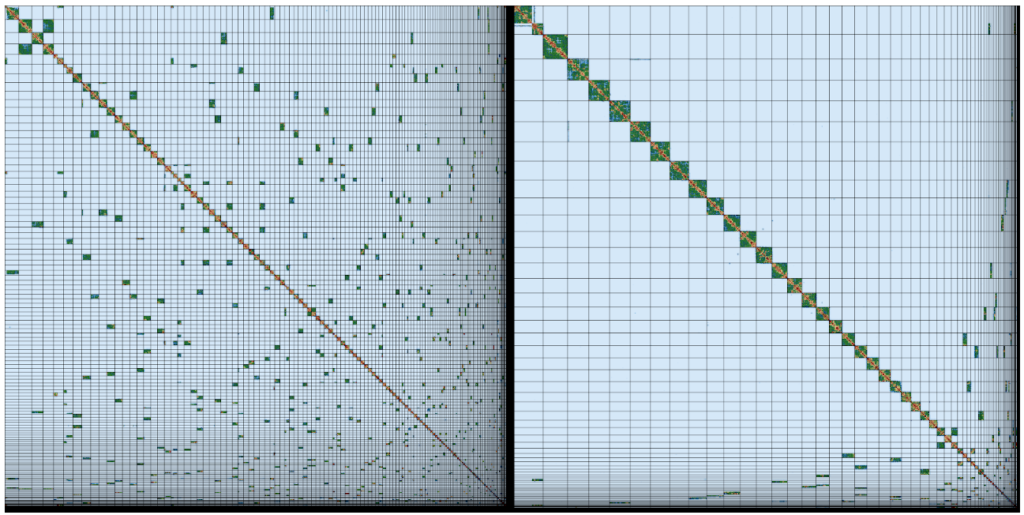
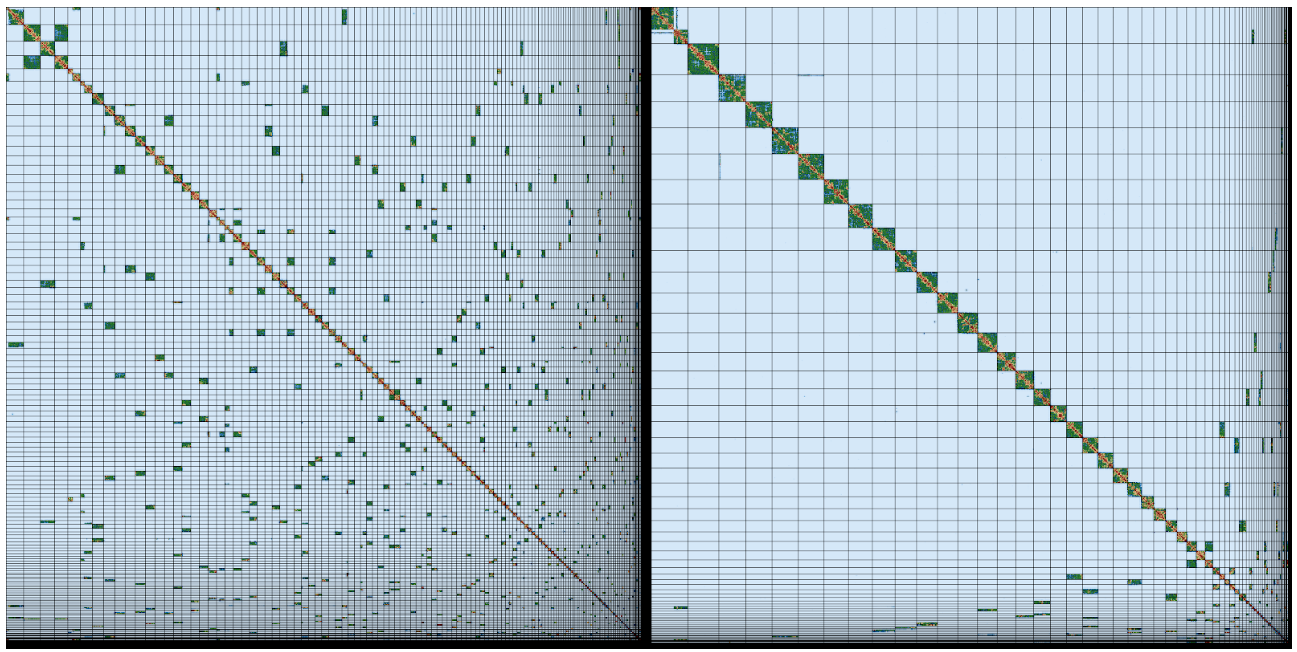
The assembly team then hands the newly-minted moth genome assembly over to Kerstin’s Genome Reference Informatics Team (GRIT: Kerstin, Joanna, Sarah, Ying, James, William, Jonathan, Alan, Damon). For the moth, Ying stress-tests the assembly with a battery of analyses, basically asking “Is this the best we can do?”. The results get handed over to Sarah, who blesses the unproblematic majority of the assembly, affirms some correct guesses, fixes the few errors and exports a quality assured assembly. James, the gatekeeper in GRIT, brokers submission of the genome assembly to the European Nucleotide Archive, part of the International Nucleotide Sequence Database Consortium, and presses the “release” button.
The new moth genome emerges into the light of a new digital day, one of 1000 species of all kinds we will extract, sequence and assemble this year. To publish the genome and announce its availability to the community to use and analyse, we write a brief Genome Note for rapid publication in Wellcome Open Research (2). Nancy marks the genome “complete”.
Now for the next one.